

Abstract
Introduction. Inhibition of Bromodomain and extraterminal (BET) proteins was effective against different acute myeloid leukemia (AML) subtypes in preclinical studies (Dawson et al. Nature 2011; Zuber et al. Nature 2011; Dawson et al. Leukemia 2013; Chen et al. Cancer Cell 2014; Gröschel et al. Cell 2014; Zhao et al. Cell Reports 2016). However, the drug had limited clinical activity, suggesting the need of ad hoc combination therapies able to target leukemia stem cells (LSCs) in their microenvironment. Hypoxia is an integral component of the bone marrow microenvironment and plays a crucial role in survival and chemoresistance of LSCs.
Aims. The study aims to elucidate the consequences of BETi treatment in AML under hypoxic conditions and identify novel potential combination strategies.
Methods. AML cell lines (OCI-AML3: NPM1- and DNMT3A-mutated, Kasumi-1: t(8;21), HL60: MYC-amplified, MOLM-13, NOMO-1: MLL-driven, KG-1: TP53-mutated) were treated with the BET inhibitor (i) GSK1215101A (250/500 nM, 48h) or the NRF2 activator omaveloxolone (NRF2a, 0.2-1 mM, 48h) and with the drug combination (72h) at 1% or 20% O2 concentration. Cell viability, apoptosis and cell cycle were evaluated by trypan blue dye exclusion assay, AnnexinV and PI staining, respectively. Gene expression profiling (HTA2.0, Affymetrix) was carried out on actively translated mRNAs isolated by polysome profiling after 16h of BETi treatment and on 61 primary AML. The TCGA AML dataset was analyzed on the cBioPortal. Gene expression correlation and enrichment analysis were performed by Pearson coefficient and GSEA, respectively. Kaplan-Meier survival curves were compared by Logrank test. Glutathione was quantified by mass spectrometry (Metabolon).
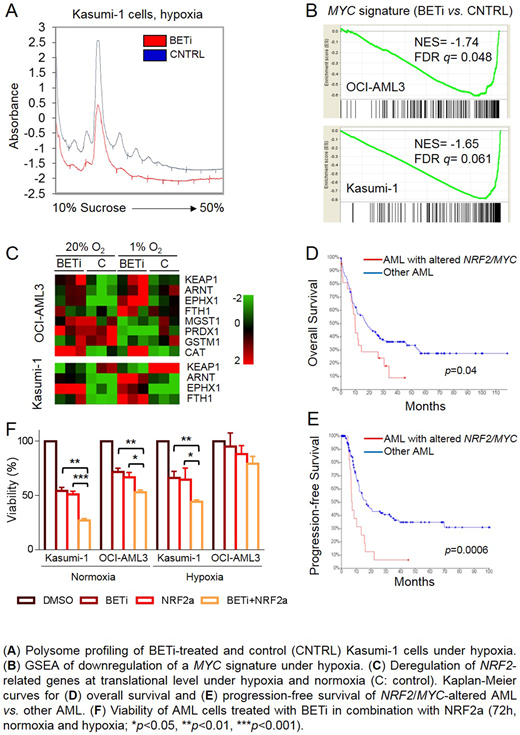
Results. BETi induced a dose-dependent reduction of cell viability in AML cells lines under hypoxia (25%-65% decrease at 500 nM) except for HL-60. Under the same conditions, the treatment caused a significant arrest in the G0/G1 phase of the cell cycle in OCI-AML3, Kasumi-1, HL-60 and KG-1 models (p<0.05) and induction of apoptosis in NOMO-1 and Kasumi-1 (40% and 50% AnnexinV+ cells, respectively, p<0.05). BETi reduced the translational rate of Kasumi-1 and OCI-AML3 cells, as determined by a decrease of disome-polysome peaks height. The treatment exacerbated hypoxia-mediated MYC suppression and associated with downregulation of a MYC signature at translational level. Moreover, it induced upregulation of the NRF2 regulator ARNT (p=0.02) and the NRF2 targets CAT, EPHX1, FTH1, GSTM1, MGST1, PRDX1 (p<0.05) under normoxia and/or hypoxia, with reduced KEAP1 mRNA and protein specifically at 1% O2 (p=0.01). These results suggest stabilization of NRF2 protein and activation of the pathway, as strengthened by increased levels of reduced and oxidized glutathione in OCI-AML3 cells (p<0.01). Based on this alternative activation of MYC and NRF2 pathway in AML, we analyzed gene expression and mutation in non-M3 AML from an internal cohort and the TCGA dataset. Upregulation of NRF2 expression and deregulation of MYC (overexpression/driver mutation) occurred in 4% and 9% of cases, respectively (independent of genomic amplification), with mutual exclusivity and an inverse correlation (p=0.03). MYC or NRF2 alterations defined a subgroup of patients with poor overall survival (10 vs. 18.1 months, p=0.04) and progression-free survival (7.2 vs. 17 months, p=0.0006). We then asked whether antioxidant gene expression was a defense response under BETi pressure. However, pharmacological inhibition of NRF2 or glutathione biosynthesis failed to potentiate the anti-leukemic effects of BETi. Conversely, activation of NRF2 pathway, which is effective as single agent on AML cells, potentiates the effects of BETi treatment in non-M3 AML, with reduced cell viability and increased apoptosis.
Conclusions. BET protein activity drives alternative NRF2 or MYC overexpression in AML, which defines a subgroup of patients with poor prognosis. NRF2 activation is finely tuned in AML, as both inhibition and activation of the pathway induce cell death. However, NRF2 activation specifically potentiates BETi treatment under hypoxia and normoxia, suggesting a novel combination therapy against AML LSCs.
Supported by: EHA Non-Clinical Junior Research Fellowship, ELN, AIL, AIRC, project Regione-Università 2010-12 (L. Bolondi), FP7 NGS-PTL project, Fondazione del Monte BO e RA project.
Cavo:Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; Amgen: Honoraria, Membership on an entity's Board of Directors or advisory committees; Adaptive Biotechnologies: Honoraria, Membership on an entity's Board of Directors or advisory committees; AbbVie: Honoraria, Membership on an entity's Board of Directors or advisory committees; GlaxoSmithKline: Honoraria, Membership on an entity's Board of Directors or advisory committees; Bristol-Myers Squibb: Honoraria, Membership on an entity's Board of Directors or advisory committees; Takeda: Honoraria, Membership on an entity's Board of Directors or advisory committees; Janssen: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau. Martinelli:Celgene: Consultancy, Speakers Bureau; Amgen: Consultancy; Pfizer: Consultancy, Speakers Bureau; Abbvie: Consultancy; Roche: Consultancy; Ariad/Incyte: Consultancy; Jazz Pharmaceuticals: Consultancy; Janssen: Consultancy; Novartis: Speakers Bureau.

